Characterised by symptoms including impaired motor and cognitive function, Huntington disease is caused by the expansion of a CAG trinucleotide repeat in the huntingtin (HTT) gene. This results in the production of an abnormal mutant HTT protein, which forms aggregates in the brain, leading to neurodegeneration and driving disease progression.
Autophagy, a mechanism employed by the cell to remove unnecessary or disfunctional components, has been reported as essential for the degradation of the mutant HTT protein aggregates. However, the degree of involvement of this process in disease progression is a controversial topic. In her first paper as senior author, Dr Karolina Pircs hopes to shed some light on this long-standing debate.
“In this paper we used a mouse model of Huntington disease, where we inject neuron specific viral vectors into the striatum to drive the over-expression of either wild type, or mutant HTT”, explains Karolina. This model exhibits a severe and rapidly developing form of mutant HTT protein accumulation. “We can tightly control many aspects of this model, making it a powerful tool for understanding the mechanisms involved in Huntington disease progression”.
A number of studies have reported that activating autophagy can reduce the amount of mutant HTT aggregates accumulating within Huntington disease neurons. In fact, these findings served as the foundation for a recent clinical trial in mild Huntington disease patients, modulating the autophagy process. “We asked whether we could reduce the frequency of mutant HTT aggregates in our disease model by boosting autophagy” explains Karolina.
After no therapeutic benefit was observed following treatment with a general ‘master regulator’ of autophagy, TFEB, the research team focused their attention on Becn1. This gene encodes a protein crucial for the formation of autophagosomes - spherical structures that engulf mutant HTT aggregates and deliver them to the lysosomes for degradation.
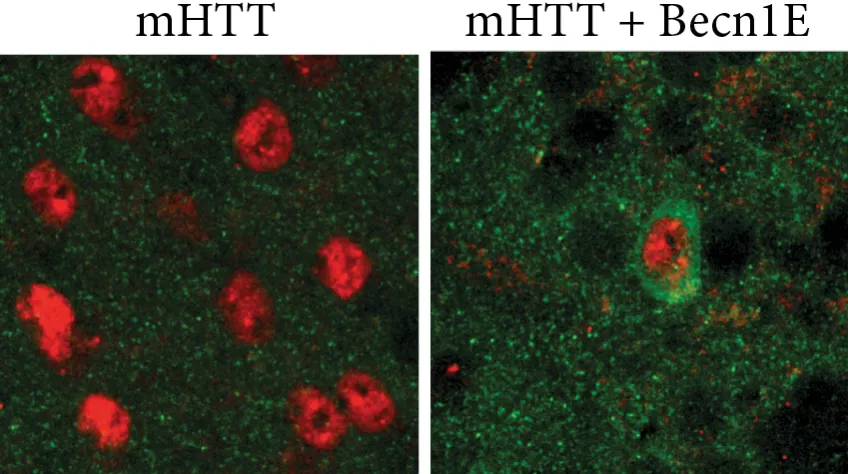
No therapeutic effect was observed upon Becn1 overexpression at a later stage, with established mutant HTT accumulation and autophagy impairments. However, when Becn1 was administrated early in the disease process - as a sort of preventative treatment - significant therapeutic efficacy was observed, with a reduction of up to half the mutant HTT aggregates.
“This study underlines the importance of understanding how autophagy is impaired in Huntington disease, as well as the time point at which it is best to take action” reflects Karolina. “Autophagy is a promising therapeutic target, but if we try to modulate it at too late stage it may be of little therapeutic value to patients”.
Publication in the journal Autophagy: Impact of differential and time-dependent autophagy activation on therapeutic efficacy in a model of Huntington disease
Contact:
Karolina Pircs, Associate Senior Lecturer.
Wallenberg Neuroscience Center, Lund University.
+46 46 222 05 44
karolina [dot] pircs [at] med [dot] lu [dot] se (karolina[dot]pircs[at]med[dot]lu[dot]se)
https://www.kpircs.com/
This research was conducted in the Molecular Neurogenetics group which is led by Prof Johan Jakobsson.
The work was supported by grants from the Swedish Research Council, the Swedish Foundation for Strategic Research, the Swedish Brain Foundation,the Swedish excellence project Basal Ganglia Disorders Linnaeus Consortium (Bagadilico), the Swedish Government Initiative for Strategic Research Areas (MultiPark & StemTherapy), the Stiftelsen Olle Engkvist Byggmästare, the Thelma Zoéga Fund for Medical Research, the Lars Hierta Memorial Foundation, the Greta and Johan Kocks Foundation, the Tore Nilsons Foundation For Medical Research, the Åhlen Foundation, the Crafoord Foundation,the Åke Wibergs Foundation, the Thorsten and Elsa Segerfalk Foundation, the Anna-Lisa Rosenberg Fund for Neurological Research, and the Royal Physiographic Society of Lund.
